# چکیده
شبکه های عصبی کانولوشن برای استخراج ویژگی هایی در سطوح abstraction
مختلف کارایی دارند. در این مقاله از این شبکه ها بر روی توالی های دنا استفاده
شده است. از بردارهای one hot
برای معرفی توالها به عنوان ورودی مدل استفاده میشود. اطلاعاتی که مربوط به جایگاه
نوکلئوتیدهای به کار رفته در این توالی ها حفظ میشود.
# مقدمه
امروزه توسط تکنولوژیهای توالییابی میتوان به سادگی رشته های دنا را خواند که
هزینه آن نیز برای یک میلیون جفت باز به طور قابل توجهی از حدود 5هزار دلار
(سپتامبر 2001) به 0.014 دلار (اکتبر2015) کاهش یافته است. همجنین میزان اطلاعات
مربوط به رشته های دنا به صورت نمایی افزایش یافته است. مثلن سایز GenBank که یکی از دیتابیس های محبوب در این
حوزه است در دسامبر 2015 تا حدود 2 بیلیون جفت باز رشد کرده است. بنابراین اگر
بتوان این حجم عظیم از داده ها را در تکنولوژی های مدرن امروزی به کار گیریم
میتوان به اطلاعات مهمی از دنا دست پیدا کرد. پس از آموزش مدلهای یادگیری ماشین از روی رشته ها، میتوانیم
این مدلها را برای پیش بینی رشته های مجهول استفاده نمود. در سالهای اخیر یادگیری
عمیق معرفی شده که در زمینه ی رشته های دنا نیز کارهایی انجام گرفته است. یک مورد
مشترک در تمامی تحقیقات مربوطه اینست که در آنها از خصیصه های خبره ها استفاده شده
بود که در اینصورت اطلاعات مهمی که مرتبط با مکان هر نوکلئوتید در رشته ها است در
نظر گرفته نشده اند که باعث افت کارایی مدلها خواهند شد. چرا از خود رشته ها
بعنوان ورودی مداهای یادگیری عمیق استفاده نشود؟ این مشکل مربوط به این مورد است
که مدلهای یادگیری عمیق بردار ها و یا ماتریس های عددی را دریافت میکنند و رشته
هایی شامل حروفی متوالی با هم را به عنوان ورودی نمیپذیرند.
در این تحقیق یک مدل یادگیری عمیق که برای بازنمایی توالیها
از بردارهای one-hot استفاده میکند و
اطلاعات مکانی هر نوکلئوتید را در خود حفظ میکند به کار گرفته شده است. در این مدل
از یک مدلی که برای دسته بندی متون به کار گرفته میشود الهام گرفته شده است که در ادامه به معرفی آن خواهیم پرداخت.
# شرح روش ها و مدل ها
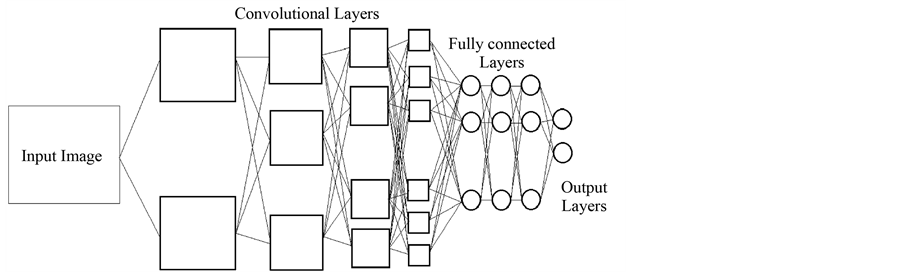
در شکل بالا یک نمونه از شبکه های عصبی کانولوشن را مشاهده میکنید که برای
تشخیص چهره به کار میرود. در این شبکه 4 لایه کانولوشن به کار رفته که هر یک از
آنها با یک sub-sampling همراه هستند و در لایه های بعد 3
لایه ی تماما متصل به کار رفته و در انتها یک لایه خروجی استفاده شده است. در لایه
های ابتدایی اشکال ساده نظیر خطوط افقی و عمودی و مورب را بدست می آورد و رفته
رفته در لایه های بعدی اشکال پیچیده تر را میتواند تشخیص دهد و از ورودی ها
استخراج کند تا جایی که در لایه کانولوشن آخر قادر به تشخیص چهره انسان خواهد بود.
از شبکه های عصبی پیچشی در متن ها کاربردهایی نظیر دسته بندی موضوعات، تشخیص
اسپم ها و دسته بندی معنایی را دارد. در متن ها برخلاف تصاویر که بصورت آرایه دو
بعدی عددی در شبکه به کار برده میشوند با بردارهایی تک بعدی از کلمات سرو کار
داریم. بنابراین نیاز به نگاشت آنها به اعداد داریم. که برای این امر میتوان از
جداول lookup استفاده نمود که در آن هر کلمه از دیکشنری را به یک بردار حرف
تبدیل میکنند. اندازه این بردارها در هر مدلی ثابت در نظر گرفته میشوند . مقادیر
آنها را از طریق شبکه های عصبی پیچشی یا متدهای دیگری نظیر word2vec
بدست می آورند. به این ترتیب با استقاده از جداول lookup اطلاعات unigramی از متن را میتوان استخراج نمود.
برای استخراج ویژگیهایی در حد bi-gram یا n-gram
ها میتوان رویکرد دیگری را به کار گرفت. به این صورت که از بردارهایone hot
برای بازنمایی کلمات استفاده میشود. سپس با اتصال بردار حروف از حروف متوالی
میتوان اطلاعات n-gramی از متن را نیز استخراج نمود.
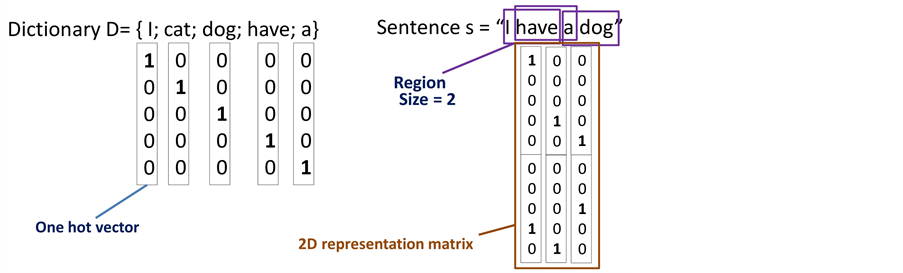
طریقه ی استفاده از بردار های one hot در شکل بالا
نشان داده شده است. فرض کنید دیکشنری ما شامل تنها 5 کلمه ی {I,cat,dog,have,a} باشد. هر یک از این کلمات را با یک
بردار one hot نمایش میدهیم. در شکل بالا این بردار مختص به هر کلمه را زیر آن
کلمه نوشته ایم. اما قبل از بدست آوردن ماتریس بازنمایی متن باید اندازه ی ناحیه
همسایگی را مشخص کنیم. در این مثال اندازه این ناحیه را برابر با 2 در نظر گرفته
ایم. به این معنی است که اطلاعات مربوط به هر دو کلمه متوالی را در کنار یکدیگر
قرار میدهیم که این اطلاعات ستونهای ماتریس بازنمایی متن را تشکیل خواهند داد. بنابراین
در انتها اگر در متنمان W کلمه داشته باشیم، W-1
ستون در ماتریس بازنمایی خواهیم داشت. این ماتریس ها را بعنوان ورودی شبکه عصبی
پیچشی به کار خواهیم برد.
حال برای استفاده از شبکه پیچشی در توالیهای دنا باید درنظر داشته باشیم که
این توالیها شامل حروفی به هم چسبیده هستند که بدون فاصله در کنار یکدیگر قرار
گرفته اند. میخواهیم همان رویکرد مربوط به متنها را در اینجا نیز به کار ببریم
بدون آنکه اطلاعات مربوط به جایگاه های نوکلئوتید ها را از دست بدهیم.

در شکل بالا نحوه ی تبدیل یک رشته ی دنا به یک توالی از کلمات (متن) را نشان
داده ایم. برای این کار از پنجره ی لغزانی با اندازه ی 3 را در نظر گرفته ایم که
با گام 1 روی رشته حرکت میکند و بخشی از رشته را که داخل پنجره قرار میگیرد را به
عنوان کلمات مورد نظرمان در نظر میگیریم. بنابراین در انتها درصورتیکه رشتهی
اولیه شامل n حرف باشد، یک متن با n-2
کلمه در اختیار خواهیم داشت. که در ادامه میتوانیم این متن را بعنوان ورودی شبکه
پیچشی خود به کار بریم که نحوه ی انجام این کار در بالا توضیح داده شد. همانطور که
میدانیم حروف به کار رفته در رشته های دنا تنها شامل جهار حرف هستند بنابراین با
در نظر گرفتن کلمات سه حرفی، تعداد حالات مختلف برای کلمات 64 حالت خواهد بود(43
حالت). بنابراین همانطور که در شکل زیر نیز نشان داده شده است اندازه ی بردارهای one hot ما در این مثال برابر 64 خواهد بود.

# دیتاست
برای ارزیابی کارایی مدل پیشنهادی از 12 دیتاست استفاده شده است.
در 10 دیتاست اول که در کار [1] به کار رفته اند، در هر دیتاست نمونه ها رشته
هایی به طول 500 جفت باز هست که در دو کلاس مثبت یا منفی قرار میگیرند. نمونه های کلاس
مثبت دارای نواحیی هستند که به دور پروتئین هیستون پیچیده اند. در حالیکه نمونه
های کلاس منفی دارای این نواحی نیستند. بنابراین اگر بتوانیم هیستون ها را از روی
توالی ها پیش بینی کنیم قادر به تشخیص الگوی بیان ژن ها خواهیم بود. این دیتاست ها
راجع به رشته های دنایی هستند که به دور پروتئینهای هیستون پیچیده اند. این
مکانیزم برای بسته بندی و ذخیره سازی رشتهای دنای بلند داخل یک نوکلئوس سلول
هستند. نامگذاری این دیتاست ها به این صورت است:
موارد H3 یا H4 نوع هیستون را مشخص میکنند. Kو یک عددی که بدنبال آن می آید نشان دهنده ی
آمینواسید تغییر یافته هستند.(مثلا K14 بیانگر اینست که 14مین آمینواسید K
دچار تغییر شده است.) ac یا me
که نوع تغییرات (acetylation یا methylation)
را نشان میدهد و عددی که به دنبال me می آید بیانگر تعداد رخداد این
تغییرات است.
در این تحقیق برای مقایسه با کارهای پیشین، در ده دیتاست اول که در کار [1]
نیز از انها استفاده شده و در همان کار ویژگیهای 4-merی استفاده شده است و از کلاسیفایر svm با کرنل RBF برای دسته بندی داده ها
استفاده شده است. همچنین برای بهبود کارایی کلاسیفایر خود از یک روش "انتخاب
ویژگی" نیز استفاده کرده اند.
دو دیتاست دیگر
دیتاستهای Splice و Promoterاز دیتاست های بنچمارک uci
هستند. دیتاست Promoter راجع به Promoter ها
میباشد. این دیتا ست شامل رشته هایی به طول 57 جفت باز هستند که در دو کلاس مثبت و
منقی قزار میگیرند. که در صورتیکه بتوانیم پیش بینی روی رشته ها بتوانیم انجام
دهیم یعنی قادر به تشخیص بیان ژن ها خواهیم بود. بهترین نتایج مربوط به این دیتاست
در کارهای پیشین مربوط به کار [2] میباشد که در کار خود از یک شبکه عصبی Knowledge-based بهره بده اند. دیتاستSplice راجع به splice junction ها میباشد.در ژن ها نواحیی که طی فرآیند رونویسی رنا حذف میشوند
اینترون نامیده میشوند و نواحی که در تولید mrna به کار میروند اکسترون نامیده میشوند. محل اتصال بین این نواحی را
splice junction گویند. این اتصالات به دو نوع exon-intron و intron-exon وجود دارند. در این
دیتاست نمونه های رشته هایی به طول 60 جفت باز هستند و به سه نوع زیر تقسیم بندی
میشوند:
EI: نمونه هایی دارای نواحی با اتصالات Exon-Intron
IE: نمونه هایی دارای نواحی با اتصالات Intron-Exon
N: نمونه هایی که در دو دسته بالا قرار
نمیگیرند.
بهترین نتایج مربوط
به این دیتاست در کارهای پیشین مربوط به کار [3] میباشد که در این
کار برای دسته بندی داده ها از c4.5 استفاده شده است.
# مدل به کار گرفته شده
در مدل به کار رفته
در این مقاله، 2لایه کانولوشن که هر یک با یک لایه subsampling
همراه هستند به کار رفته اند که این لایه ها برای استخراج ویژگیها از روی ماتریس
های بازنمایی بکار میروند. در ادامه این ویژگیها وارد یک لایه تماما متصل و دارای
100 نورون میشوند که در این لایه برای کاهش اثر پیش برازش از dropout
با مقدار 0.5 استفاده شده است. در انتها یک لایه خروجی softmax
برای تشخیص کلاس داده های ورودی به کار میرود.
# ارزیابی و نتایج
برای ارزیابی کارایی مدل پیشنهادی سه مرتبه از 10-fold cross validation
استفاده شده و میانگین مقادیر بدست آمده برای دقت پایانی استفاده میشود.
در مقایسه با بهترین کارهای پیشین مربوط به هر دیتاست همانطور که در جدول زیر
نیز مشاهده میکنید، در تمامی دیتاست ها همواره شاهد بهبود دقت بوده ایم. تفاوت دقت
در کمترین میزان 1 درصد و بیشترین میزان آن بیش از 6 درصد میباشد.

# مراجع
1. Higashihara, M., Rebolledo-Mendez, J.D., Yamada, Y. and Satou, K. (2008) Application of a Feature Selection Method to Nucleosome Data: Accuracy Improvement and Comparison with Other Methods. WSEAS Transactions on Biology and Biomedicine, 5, 153-162.
2. Towell, G., Shavlik, J. and Noordewier, M. (1990) Refinement of Approximate Domain Theories by Knowledge-Based Artificial Neural Networks. Proceedings of the 8th National Conference on Artificial Intelligence, Boston, 29 July-3 August 1990, 861-866.
3. Li, J. and Wong, L. (2003) Using Rules to Analyse Bio-Medical Data: A Comparison between C4.5 and PCL. Proceedings of Advances in Web-Age Information Management 4th International Conference, Chengdu, 17-19 August 2003, 254-265.